
In this section, we will show how to define coding regions, so that Aligner can correctly describe the amino acid changes caused by mutations. Since this is typically done when a reference sequence is known, we will also show how to use a reference sequence for mutation detection.
The reference sequence is typically a known sequence from a text file (Aligner can read plain text files, FASTA files, and Genbank formated files). For our example, we will export a consensus sequence, and then re-import it into a new project, where we make it the reference sequence.:
We want to first add the sequence we just exported. If you saved it to your desktop, you can just drag it onto the project window; otherwise, use "Open..." in the "File" menu, and select the sequence we just exported (it is called "Contig1.fasta", unless you chose a different name).
You should now see a sequence called "Contig1" in the "Unassembled Samples" folder. The name could be confusing later, so we'll rename it:
In the dialog that comes up, rename the sequence to "MyReference":
Click "OK", and you'll see the name change in the project view. Note that renaming the sequence did not make it a reference sequence! We did this just to we can easily identify it in the project view. We'll tell Aligner to use this sequence as the reference sequence in the next step.
You should see that the icon used in the project view changes, and the description changes from "Text" to "Reference".
At this point, it is a good idea to save the project. If this was one of your own projects, you may want to save a copy of the project that just contains the reference sequence using "Save As...", so you can re-use it later for other sequences.
Next, we want to add the sequence traces from the PolyPhred example project:
Aligner import all 10 files in this folder (it may ask you first if you really want to do that, depending on your warning preferences). Your "Unassembled Samples" folder now contains 11 samples. Click on the "Unassembled Samples" folder, then choose "Align to Reference Sequence" from the "Contig" menu. The alignment should produce one contig with 4 traces and the reference sequence, leaving 6 samples in the "Unassembled Samples" folder.
Select the contig in the project view, then choose "Find Mutations" from the "Contig" menu. Look at the result table - notice that the numbers in the "Start" column and the base number in the "Content" column are the same. We'll change this next by defining the coding region in the next section.
To tell Aligner where in your coding regions are in your reference sequence, you need to add a "codingSequence" tag to each exon. You can add these tags to the consensus sequence or to the reference sequence; in general, it is better to add them to the reference sequence.
Aligner will use codingSequence tags from the reference sequence only if the reference sequence is also used as the consensus sequence. So first, we want to make sure this is the case:
For this exercise, we will pretend that this reference sequence contains two exons somewhere in the middle of a gene that we are analyzing. We will first add two "codingSequence" tags that describe where the exons are. Then, we will add a "codonStart" tag to the first exon, which allows us to do two things: we can specify where the first complete codon in this exons starts (at the first, second, or third base), and we can specify what the number of the nucleotide at this position is. Let us assume the following gene structure:
Open a contig view for our contig by double-clicking on Contig1 in the project view. Then, click on the any base in our reference sequence to select it. Now go to base 185; the fastest way is to select "Base Number..." from the "Go" menu. In the "Go To Base" dialog, type "185", then press "OK". Aligner will now go to the first start codon at base 185.
We want to define the coding region to start here, and extend to base 220. With the base 185 still selected, go to the "Sample" menu, and choose "Tag -> Add Tag...". In the "Add Tag" dialog, first click on the pull-down menu at the top, and select "codingSequence" as the tag type. Then, enterthe number 220 in the "End:" text box. Your tag dialog now should look like this:
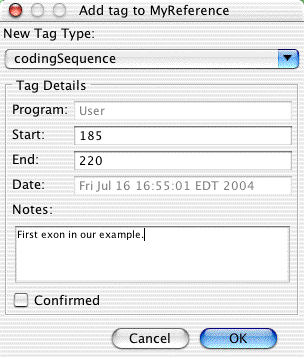
You can also add any text in the "Notes:" section, if you like. Then click "OK".
We add the second codingSequence tag the same way, except that this time, we won't bother to first go to the base where the coding region starts. Just right-click (OS X: control-click) on any base in the "MyReference" sequence, and choose "Add Tag..." from the popup menu. In the "Add Tag" dialog, again choose "codingSequence" as the tag type. Then enter "375" in the "Start:" field, and "9999" in the end field (if you enter a number that extends beyond the end of the sequence, Aligner will replace it with the number of the last base when you click "OK"). Your dialog should now look like this:

Click "OK". If we would do the mutation detection now (you can try it), Aligner would take the first base in the first coding regions, base 185, as the start of the first codon, and give it the base number 1. But when you are analyzing exons somewhere in the middle of a gene, the first base may not be the start of the first codon; and you probably will want to have the correct numbering, too. We can do both of these things by adding a "codonStart" tag:
Your dialog now should look like the one below:

Click "OK" to save the changes. Now, we are ready to find mutations again. In the project view, select the contig, and then choose "Find Mutations" from the "Contig" menu (or from the popup menu).
Tip: If you are often comparing different samples to the same
reference sequence, you can save time by creating a project that contains only
the reference sequence. Mark your reference sequence as the reference sequence,
and add all the necessary codingSequence tags, as described above. Save this
project, and save a copy or
two
under
a different
name using
"Save
Project
As..." .
When you want to analyze at a new set of samples, open the project with the
reference sequence, and right away save it under a different name, using "Save
Project
As...". Then, add the samples you want to analyze. Your reference sequence
is already setup, so no need to add the tags again.
In the future, we plan to add the ability to save project templates (stationaries),
which will simplify this process a bit.
When Aligner is done, look at the result table:

Note that the numbers in the "Content" section have changed - they now are relative to the start of the coding sequence. Mutations in intron regions will annotated using the "n-x" or "m+x" conventions, depending on how close they are to the start of the next exon (n) or the end of the previous exon (m), with x being the distance.
As we said above, you can define the coding sequence by adding tags to the consensus sequence or to the reference sequence. Usually, you should add the tags to the reference sequence - but keep in mind that the coding region tags from the reference sequence will be only be used if:
When adding "codonStart" tags, there are also a few things to keep in mind:
When working with your own data, you can import the reference sequence from Genbank-formated files. Aligner will use the "CDS" line in Genbank files to create a corresponding "codingSequence" tag (but only if the CDS tag described just a single region in forward direction - more complicated CDS lines are not yet handled, and will be ignored). In addition to the "CDS" line, Aligner will also read the "codon_start" line and add a corresponding "codonStart" tag (however, you would still need specify the base number).
Note that the tag content does not get updated when you add or change tags, or even make edits that change the consensus sequence. To update the tags, you'll need to find mutations again (which will remove the old tags, and add new ones ...).
The next section shows how to exclude regions from mutation analysis, which can be useful to work around artifacts in sequence traces, or to limit the search for mutations to specific areas.
Aligner Home Page - Quick Tour Start - Previous - Next: Specifying Where To Find SNPs