This section of the quick tour describes some of the basic editing functions, like changing, adding, and deleting bases, removing reads from contigs, and splitting and merging contigs.
Before we start, two general notes:
Open the "Example1" project in the CodonCode Aligner Examples folder. If all the samples are in the "Unassembled Samples" folder (for example because you are using Aligner in Demo mode and therefore could not save your work in the previous sections), then go ahead and clip the ends for all seven samples, and then assemble them. Double-click on the resulting contig to open the contig view.
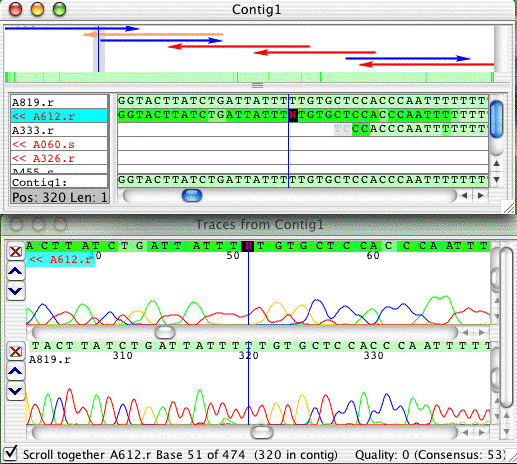
Go to base 320 in sample "A612.r", which is an N. Double-click on the consensus base at this position to open the trace view for the two samples at this position. Her is what this section looks like:
Select the N in the contig view or the trace view, and press the "T" key on the keyboard. Now it looks like this:

Note that the N is replace by a lower case "t", and the cursor is moved to the next base. If you select the "t" that you just edited in the trace view, you will notice that Aligner has assigned the quality value of 98 to the base. Quality values of 98 mean "low confidence edit". If you press the shift key while typing a base, you will notice that the letter now is an upper case letter, and that the quality value is 99, meaning "high confidence edit".
If you are curious: when calculating the consensus sequence quality, Aligned will substitute the edit quality values with numbers that you can set in the "Consensus method" preferences. The default qualities used are 10 for a low confidence edit, and 30 for a high confidence edit. You should use high confidence edits sparingly, if at all, since they can cause problems later, for example when assembling with Phrap (Phrap puts reads that differ at high confidence edits into separate contigs).
You can undo simple edits like the one we just did by selected "Undo" from the "Edit" menu. The "Undo" menu item will show you what you are undoing - in our example here, it says "Undo Base Edit". After undoing, you can re-do the edit by selecting "Redo Base Edit" from the "Edit" menu. You can also use the keyboard shortcuts for undoing (Command-Z on OS X, Control-Z on Windows) and re-doing (Command-Y on OS X, Control-Y on Windows).
You will be able to undo and redo multiple edits, but there are a few things to keep in mind:
Now that you learned how to use undo and redo, try selecting several bases in the contig view or trace view, and then replace them with a different base. What happens?
To delete a base, select the base in the contig or trace view, go to the "Sample" menu, select "Delete", and choose "Selection - Fill from Left" or "Selection - Fill from Right". Try is out and see what happens (for samples that are not part of contigs, both actions do exactly the same). You can also use the "Delete" and "Backspace" keys on Windows, and the "delete" and "del" keys on OS X; try it out!
When you delete bases manually, the part of the sequence before or after the deletion point will probably be mis-aligned. Depending on the local coverage and the quality of the samples, you may or may not change the consensus sequence.For now, use "Undo Deletion" from the "Edit" menu to undo the changes you just made.
Deleting all bases from the cursor to the start or end of the sample is also often useful, for example if your projects really don't allow you to use Aligner's end clipping. One way to do this is go to the "Sample" menu, and select "Delete > From Sample Start" or "Delete > To Sample End". Another way is to press "shift-home" (keep the shift key pressed while pressing "home"), which will select all bases from the cursor to the beginning of the sample, or "shift-end" to select to the end of the sample. Then, press the appropriate delete or backspace key (tip: try one of the delete/del/backspace keys - if it was the wrong one, just undo the deletion by pressing control-Z (command-Z on OS X), and use the other one).
There is also a similar "Delete" submenu in the "Contig" menu, which allows you to delete all bases in all samples to the start or the end of a contig. You need to have a contig base selected to be able to use these menu items.
To insert a new gap in a sample, select the base after the insertion point, and then press the space bar, or press shift-space (keep the shift key pressed while pressing the space bar). Pressing the space bar will move all bases in the sample after the insertion to the right; if you keep the shift key pressed while pressing the space bar, all bases before the insertion point will be moved to the left instead. Hard to explain, easier to see - just try it out on a sequence that is in the middle of our contig. Instead of using the space key or shift-space, you can also use the "Insert Gap" manu items in the "Sample" menu.
To insert a new base, you need to first insert a gap character, and then change it to the base you want to insert. If you really need to do this a lot and find this too inconvenient, please let us know!
To move gaps in sequence, select the gap in the contig view, and then choose "Move Gap Left" or "Move Gap Right" from the "Sample" menu. Or, better, select the gap and use the keyboard shortcuts: on Windows, use alt-F5 to move a gap left, and alt-F6 to move a gap right; on OS X, use option-F5 to move a gap left, and option-F6 to move a gap to the right. Try it!
This section describes the most common actions to do with contigs - adding sample to and removing samples from contigs, splitting contigs, and merging contigs.
To unassemble a contig, select the contig in the project view, and then choose "Unassemble" from the "Contig" menu. All samples in the contig will be moved to the "Unassembled Samples" folder, and all gaps in these samples will be removed. Try it; then select the "Unassembled Samples" folder in the project view, and assemble the contig again.
To remove samples from contigs, first select the sample or samples in the contig view. Then, go to the "Edit" menu, and choose either "Move to Unassembled Samples" or "Move to Trash", depending on whether or not you plan to re-use the samples later (samples in the trash are not lost, you can later move samples from the trash back to the "Unassembled Samples" folder).
You can also go the the project view, select the sample you want to move, and drag it to the trash or the "Unassembled Samples" folder.
You will be able to remove samples at the beginning or end of contigs, or samples in regions that are covered by other samples. If you try to remove a sample in the middle of a contig so that removing the sample would create a hole in the contig because no other sample covers this region, you will see a warning like this:

If splitting the contig into two pieces is what you wanted to do, go on to the next section!
To split a contig into two halves, first select the positions where you want to split it in the contig view. Open the contig view for the contig in the "Example1" project. Click on any consensus base, then select "Base Number..." from the "Go" menu. Type 700 in the"Got to" dialog that appears, then press the "Go To" button. This will move the cursor to consensus base 700. Your contig view should look like this:
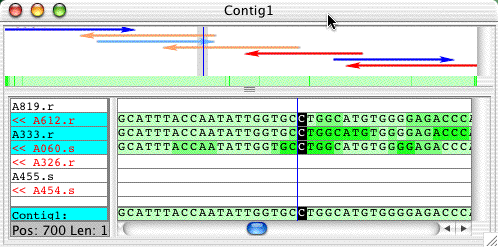
Now, go to the "Contig" menu, and select "Split Contig". Aligner wil close the contig view, and now show two contigs in the project view:

The initial contig was split at the cursor position by putting the three reads that had all or most bases before the cursor into the new contig "Contig1_left", and the four reads that had all or most bases after the cursor position into the new contig "Contig1_right".
You can merge pre-existing contigs in Aligner, and add new reads to contigs, by selecting the contigs and reads in the project view, and the choosing "Assemble" from the "Contig" menu. Try this by selecting the two contigs you just created, and assembling them - you will end up with one contig again.
Alternatively, you can add reads and contigs to another contigs by drag and drop in the project view:
When you drop samples or contigs onto an existing contig, Aligner will start an assemble of the contig and the dropped samples and contigs, and you will see a progress dialog (if the target contig is an alignment to a reference sequence, Aligner will actually to an alignment, not an assembly). Sample and contigs that satisfy the current assembly (or alignment) criteria will be added to the contig; samples that do not meet the minimum criteria will remain where they were.
You can use the interactive features we just looked at to manually correct the assembly of difficult regions like repeats or rRNA clusters, where the initial assembly generated by CodonCode Aligner or Phrap contains misassemblies.
Aligner Home Page - Quick Tour Start - Previous - Next: Copying sequences