Phylogenetic Trees in CodonCode Aligner
CodonCode Aligner supports building Neighbor-Joining trees for your contigs. The trees are displayed right next to your samples in the contig view and they automatically sort your sequences by differences. View your trees with branch lengths that represent the distance between your samples, or see the topolgy only. Phylogenetic trees can be exported in Newick format.
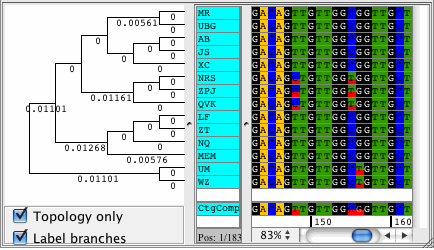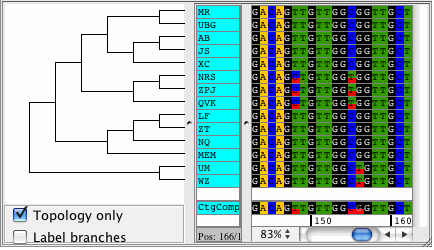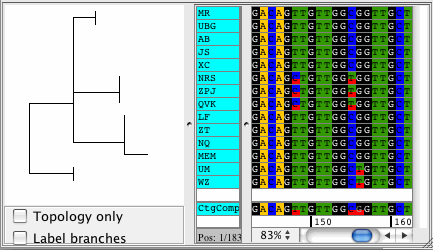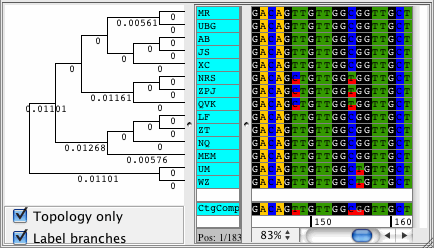
Popular uses for the Neighbor-Joining trees in CodonCode Aligner are to build trees to see how much your sequences differ, for quailty control, and to sort your sequences by distance.
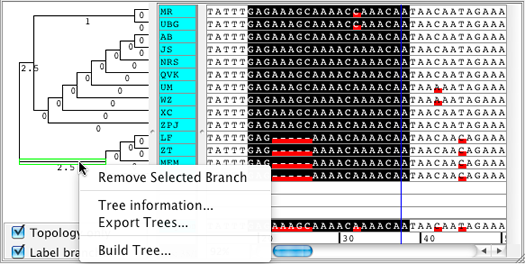
Phylogenetic trees can also be built for selected bases only (instead of for the whole contig). Use this feature to sort your samples by similarity for only a specific region of your contig.
If you have a phylogenetic tree for your contig, you can split the contig using the tree. For example if you want a separate contig for a part of the tree where the samples have many differences compared to the rest, you can use this feature to split the original contig into 2 new contigs.
Here is an example of a tree that was built for the selected bases only. The tree is then used to split the contig by removing the selected tree branch: