Sequence Alignment with CodonCode Aligner
CodonCode Aligner supports two common uses of sequence alignments: alignments to reference sequences, and multiple sequence alignments with ClustalW, MUSCLE, or built-in alignment methods. Alignments can be edited in CodonCode Aligner, and exported in commonly used format like NEXUS/PAUP and Phylip.
Align to Reference Sequences
Align your sequences to reference sequences you designate. CodonCode Aligner lets you designate multiple reference sequences, and will automatically pick the best reference sequence for each sample.
Use reference sequence alignments to:
- Find differences to known sequences.
- cDNA to genomic DNA alignments.
- Combine sequences into a contig even of they do not overlap.
- Limit your analysis to the region you are interested in.
You can choose whether the reference sequence should be ignored when building the consensus, or used as the consensus sequence. When ignoring the reference sequence, you can choose which character (N, X, or '-') to use in regions without coverage to simplify downstream analysis with BLAST or other tools.
Multiple Sequence Alignment: Align Contigs and Sequences
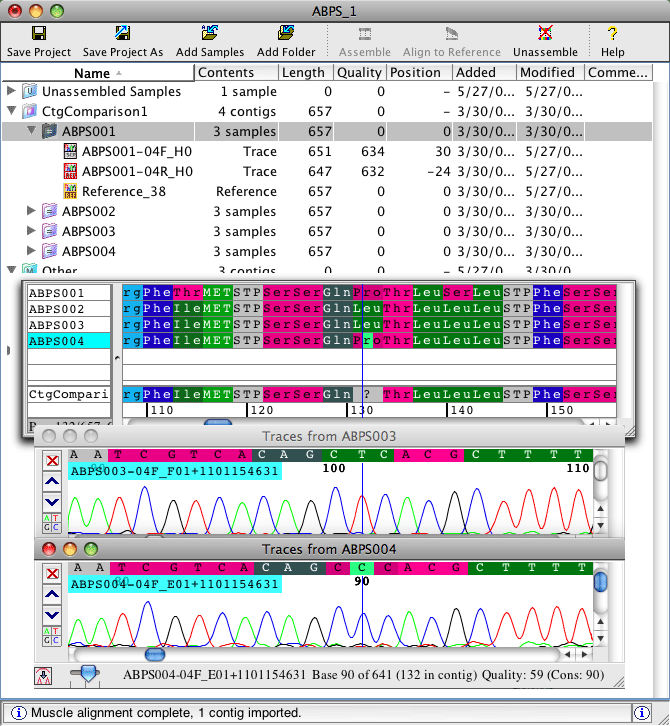
CodonCode Aligner lets you align sequences to each other with MUSCLE, ClustalW, or the built-in alignment methods. A typical use is to first assemble several sequence reads for each clone into contigs, and then align the consensus sequences for the contigs.
When you want to compare consensus sequences, most other programs force you to either duplicate the consensus sequences, or to export the sequences for alignment with a separate outside program like Clustal. To verify any differences, you then have to work with two separate copies of your sequence data.
CodonCode Aligner can greatly simplify this step:
- Align your contig sequences directly in CodonCode Aligner, while keeping the relation to the underlying sequence traces intact (to align sequences and contigs, select the sequences and contigs, then choose "Compare contigs" from the "Advanced Assembly" in the "Contig" menu).
- Go back to the sequence traces with a simple double-click.
- Just one linked copy of your data means you can edit anywhere - in the sequence traces, the consensus sequence, or the alignment of contigs. Updates in the other views will be done automatically.
- Choose between the standard multiple sequence alignment programs MUSCLE and ClustalW, or use the built-in algorithms to control parameters like minimum percent identity.
- If necessary, CodonCode Aligner will reverse-complement contigs before alignment.
You can edit your contig alignment right in CodonCode Aligner. For difficult alignments that require a lot of manual gap adjustment, CodonCode Aligner supports round trip editing: export your aligned sequences, edit in you favorite editor like Se-Al or BioEdit, and re-import the results. CodonCode Aligner will automatically update the gap placements, and retain the connection to the underlying sequence chromatograms.
An screen shot that illustrates contig alignments is shown below. For more information, watch the tutorial movies about comparing contigs and round trip editing.